
مرض الأنيميا المنجلية (Sickle cell disease) هي مجموعة من الاضطرابات الوراثية النادرة التي تصيب خلايا الدم الحمراء، بحيث يتغير شكلها، وتظهر علامات المرض على الطفل خلال العام الأول من ولادته، عادة خلال الشهر الخامس، وتختلف شدة الأعراض وتطورها من مريض لآخر، ويهدف العلاج بشكل أساسي إلى تخفيف الأعراض وحماية المريض من أي مضاعفات محتملة.
أسباب الأنيميا المنجلية
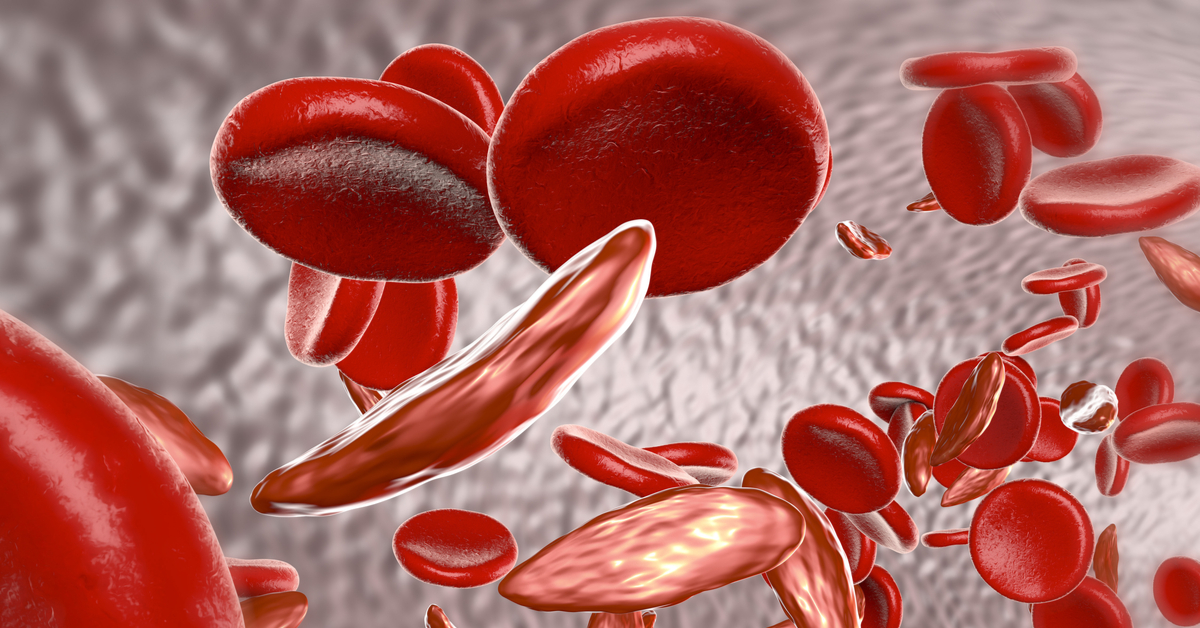
الأنيميا المنجلية هي مرض وراثي ينتقل بين العائلات، ينتج عن وراثة جين يسمى الهيموغلوبين S أو الهيموغلوبين المنجلي، بدلاً من الجين الطبيعي المسؤول عن تكوين الهيموغلوبين، والمعروف بالهيموغلوبين A، والهيموغلوبين هو المكون الأساسي لخلايا الدم الحمراء، والذي ينقل الأوكسجين من الرئتين إلى أنحاء الجسم، بينما يتسبب الهيموغلوبين المرتبط بفقر الدم المنجلي في جعل خلايا الدم الحمراء صلبة ولزجة ومشوهة.
كيف يتم توريث مرض الأنيميا المنجلية؟
تحدث الأنيميا المنجلية عندما يرث الابن زوجاً من الجينات المنجلية من كلا الوالدين، ويحدث ذلك إما من خلال أن يكون كلا الوالدين حاملين للجين المنجلي، أو أن يكون أحد الوالدين مصاباً والآخر حاملاً للجين المصاب.
مثال توضيحي في حال كان كلا الوالدين يحملان جيناً مصاباً وجيناً سليماً من الهيموغلوبين، وتكون النسب المحتملة لإصابة الأبناء كما يأتي:
- احتمال 25% أن يكون الطفل طبيعياً، بحيث يرث زوج من الجينات الطبيعية من الهيموجلوبين A.
- احتمال 50%، أن يكون الطفل حاملاً لمرض الأنيميا المنجلية، بحيث يرث جين واحد من الهيموغلوبين A الطبيعي وجين واحد من الهيموغلوبين S، وهذا الطفل لا تظهر عليه أي أعراض للأنيميا المنجلية.
- احتمال 25%، أن يكون الطفل مصاباً بالأنيميا المنجلية، بحيث يرث جينين من الهيموجلوبين S، جين من الأم وجين من الأب.
النسب المتوقعة في المثال السابق تنطبق على الإناث والذكور على حد سواء.
كيف يؤثر وراثة الجين المنجلي على خلايا الدم الحمراء الطبيعية؟
يتسبب الهيموغلوبين المنجلي كما ذكرنا سالفاً في تغيير خصائص الهيموغلوبين الطبيعي، ما يجعله غير قادر على أداء وظيفته الأساسية، ويمكن شرح آلية حدوث ذلك كالآتي:
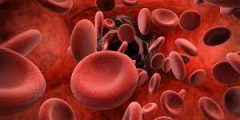
- الهيموغلوبين الطبيعي يتميز بالمرونة وسهولة الذوبان والانسياب، بينما يكون الهيموغلوبين المنجلي صلباً ولزجاً ومشوهاً، ما يعيق حركة خلايا الدم الحمراء في الأوعية الدموية، ويزيد من تكتلاتها وتجمعها، ويزيد من خطر انسداد الأوعية الدموية.
- خلايا الدم الحمراء التي تحوي الهيموغلوبين الطبيعي تعيش حوالي 120 يوماً، بينما تستمر دورة حياة خلايا الدم الحمراء المنجلية فقط من 10 إلى 20 يوماً، ما يزيد العبء على نخاع العظم لتعويض النقص في خلايا الدم الحمراء، ومع الوقت يصبح نخاع العظم غير قادر على تعويض النقص المستمر.





الشخص الذي يحمل جين واحد من الهيموغلوبين S، أو الجين المنجلي، يعد حاملاً للمرض، أو يعرف بسمة فقر الدم المنجلي (Sickle cell trait)، لا تظهر عليه أي أعراض، لكنه يستطيع توريث أبنائه المرض.
إقرأ أيضا:أسباب وعلاج فقر الدم: دليل شامل